2026
3'-Phosphorofluoridates for P(V)-Based Oligonucleotide Synthesis
Oligonucleotides (ONs) are fundamental to modern life sciences and nucleic acid-based therapeutics. Although P(III)-based phosphoramidite chemistry remains industry standard for ON synthesis, its requirement for phosphorus oxidation every coupling cycle is operationally complex. Inspired by Michelson and Todd, who synthesized the first dinucleotide via a 3′-phosphorochloridate [P(V)–Cl] intermediate, we have developed a strategy for ON synthesis using nucleoside 3′-phosphorofluoridates [P(V)–F] as bench-stable building blocks. In the presence of silicon-based activators, the enhanced reactivity of the P(V)–F bond resulted in more efficient nucleotide coupling than conventional P(III)-based phosphoramidite chemistry. Furthermore, automated solid-phase synthesis of a 20-mer ON can be achieved on a standard DNA/RNA synthesizer. Our study demonstrates that P(V)-based phosphorofluoridate chemistry facilitates oxidation-free ON synthesis, providing a complementary approach to conventional P(III)-based phosphoramidite chemistry.
Nana Mihara, Syuya Inoue, Kazuho Okunishi, Yasufumi Fuchi, Yuta Ito, Yoshiyuki Hari, Tsubasa Inokuma, Noriaki Minakawa, Noriko Saito-Tarashima,* J. Am. Chem. Soc. in press (2026).
Thio-modification effects on mRNA translation using a PureCap-based capping method
Zheyu Meng, Yiwei Liu, Yuko Nakashima, Masahito Inagaki, Seigo Kimura, Yukari Kondo, Fumitaka Hashiya, Naoko Abe, Noriko Saito-Tarashima, Noriaki Minakawa, Yasuaki Kimura, Hiroshi Abe, RSC Chem. Biol. 48, in press (2026).
2025
Identification of 4'-thiouridine as an orally available antiviral agent targeting both RdRp and NiRAN functions of SARS-CoV-2 Nsp12
Minjae Kim, Myoung Kyu Lee, Inseong Jo, Yejin Jang, Soo Bong Han, Juyeon Lee, Ayeon Yang, Dnyandev B. Jarhad, Hongseok Choi, Yuhei Nogi, Noriko Saito-Tarashima, Noriaki Minakawa, Meehyein Kim, Lak Shin Jeong, J. Med. Chem. 68, 12414–12433 (2025).
Development of a Gryllus bimaculatus-Based Assay System for Evaluating Chemically Modified siRNAs
Small interfering RNAs (siRNAs) hold great therapeutic promise due to their ability to selectively silence disease-associated genes. Although chemically modified siRNAs have demonstrated clinical efficacy, their development remains hindered by challenges such as stability and delivery under physiological conditions. Furthermore, in vitro screening of chemically modified siRNAs using cultured cells is cost-effective but often fails to recapitulate in vivo complexity, limiting predictive accuracy. To address this, we have developed a transfection-free siRNA evaluation platform using Gryllus bimaculatus (Gb), an insect model with natural RNA uptake capacity. We first demonstrated that 21-nucleotide siRNAs induced RNA interference upon abdominal injection under conditions of free uptake. We then generated transgenic lines harboring an EGFP reporter fused to the therapeutic siRNA target sequence and integrated into the β-actin locus via clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9-mediated knock-in. Two transgenic strains (s1 and d1) were established and validated. We compared unmodified and chemically modified siRNAs designed using enhanced stabilization chemistry (ESC), a clinically validated modification pattern incorporating 2'-O-methyl, 2'-fluoro, and phosphorothioate modifications. While ESC-modified siRNAs showed reduced activity compared to unmodified natural siRNAs in conventional cell-based assays requiring transfection reagents, they exhibited consistent gene silencing in Gb, reflecting their enhanced biochemical stability under free uptake conditions at picomole-scale doses. These results establish Gb as a scalable, cost-effective, and biologically relevant platform for evaluating therapeutic siRNAs, particularly those incorporating chemical modifications.
Taketo Inoue, Shintaro Inoue, Yuhei Nogi, Jun Tsukimoto, Noriko Saito-Tarashima, Sumihare Noji, Taro Mito, Noriaki Minakawa, Biological and Pharmaceutical Bulletin, 2025, 48, 941.
Synthesis and resolution of 4'-substituted nucleosides with potential antiviral and antisense strategies
Yukino Endo, Kyohei Itoh, Hiroya Kan-No, Hideaki Wakamatsu, Yoshihiro Natori, Yukako Saito, Asako Kaise, Yuhei Nogi, Noriko Saito-Tarashima, Noriaki Minakawa, Yuichi Yoshimura, J. Org. Chem. 90, 2008–2021 (2025).
Synthesis of 4′-thiomodified GS-441524, a nucleoside unit of Remdesivir, as an anti-SARS-CoV-2 agent
The COVID-19 pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), created an urgent need for effective antiviral treatments. While Remdesivir (GS-5734) and its parent nucleoside GS-441524 have been explored as anti-SARS-CoV-2 agents, their use is limited by toxicity concerns. Here we synthesized a 4′-thiomodified derivative of GS-441524 (1β) using a refined C-glycosylation strategy. In VeroE6 cells infected with SARS-CoV-2, 1β demonstrated modest antiviral activity without detectable cytotoxicity. Although its potency was lower than Remdesivir, its favorable safety profile suggests 1β has potential as a safer antiviral for RNA viruses.
Nana Mihara , Yuhei Nogi , Noriko Saito-Tarashima , Takaaki Koma , Masako Nomaguchi , Noriaki Minakawa, Chemistry Letters, 2025, 54, upaf080.
3-Deazaguanosine inhibits SARS-CoV-2 viral replication and reduces the risk of COVID-19 pneumonia in hamster
The COVID-19 pandemic highlighted the serious threat that coronaviruses have on public health. Because coronavirus continuously undergoes cross-species transmission, additional therapeutic agents and targets are urgently needed. Here, we show that a 3-deazapurine ribonucleoside, 3-Deazaguanosine (C3Guo, 2), has potent antiviral activity against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Unexpectedly, C3Guo (2) does not act as an inhibitor of RNA-dependent RNA polymerase (RdRp), which is the therapeutic target of two key nucleoside/nucleotide inhibitors approved for the treatment of COVID-19 (Remdesivir and Molnupiravir); instead, it seems to function by targeting the capping machinery of viral RNA. In hamsters infected with SARS-CoV-2, administration of 2 markedly reduced infectious viral titers, and prevented the development of COVID-19 pneumonia better than Molnupiravir. The potency of 2 against SARS-CoV-2 underscores its potential as an effective therapeutic agent for COVID-19 and future zoonotic coronavirus infections and raises the possibility of antiviral nucleoside analogs with alternative therapeutic targets to RdRp.
Noriko Saito-Tarashima, Takaaki Koma, Naoto Hinotani, Keigo Yoshida, Moka Ogasa, Akiho Murai, Syuya Inoue, Tomoyuki Kondo, Naoya Doi, Koichi Tsuneyama, Masako Nomaguchi, Noriaki Minakawa, 2025, 28, 112140.
2023
Synthesis and Behavior of DNA Oligomers Containing the
Ambiguous Z-Nucleobase 5-Aminoimidazole-4-carboxamide
5-Amino-1-β-D-ribofuranosylimidazole-4-carboxamide 50-monophosphate (ZMP) is a central intermediate in de novo purine nucleotide biosynthesis. Its nucleobase moiety, 5-aminoimidazole-4-carboxamide (Z-base), is considered an ambiguous base that can pair with any canonical base owing to the rotatable nature of its 5-carboxamide group. This idea of ambiguous base pairing due to free rotation of the carboxamide has been applied to designing mutagenic antiviral nucleosides, such as ribavirin and T-705. However, the ambiguous base-pairing ability of Z-base has not been elucidated, because the synthesis of Z-base-containing oligomers is problematic. Herein, we propose a practical method for the synthesis of Z-base-containing DNA oligomers based on the ring-opening reaction of an N1-dinitrophenylhypoxanthine (HxaDNP) base. Thermal denaturation studies of the resulting oligomers revealed that the Z-base behaves physiologically as an A-like nucleobase, preferentially forming pairs with T. We tested the behavior of Z-base-containing DNA oligomers
in enzyme-catalyzed reactions: in single nucleotide insertion, Klenow fragment DNA polymerase recognized Z-base as an A-like analog and incorporated dTTP as a complementary nucleotide to Z-base in the DNA template; in PCR amplification, Taq DNA polymerase similarly incorporated dTTP as a complementary nucleotide to Z-base. Our findings will contribute to the development of new mutagenic antiviral nucleoside analogs.
Yuhei Nogi et al., Molecules 2023, 28, 3265.
Chemistry of cyclic dinucleotides and analogs
Cyclic dinucleotides (CDNs) are a class of macrocyclic natural product consisting of two nucleotides linked via two phosphodiester linkages. For example, a CDN composed of two guanine bases is called cyclic-di-GMP (c-di-GMP). CDNs are biosynthesized from two nucleoside 5′-triphosphates by cyclase enzymes, such as guanylate cyclase which synthesizes c-di-GMP. Since the 1980s, as the biological significance of CDNs has become clear, chemists have focused on the chemical synthesis of CDNs and analogs. These efforts have contributed significantly to the elucidation of the diverse biological process regulated by CDNs. This chapter addresses the chemistry of CDNs and analogs. In the first half of this chapter, synthetic strategies for making CDNs are outlined. The strategies are classified according to the key reactions which form the internal phosphodiester linkages and construct the CDN skeleton, namely dimerization and cyclization. In the second half of the report, chemically modified CDN analogs developed using the above chemistry are introduced.
Noriko Saito–Tarashima et al., Handbook of Chemical Biology of Nucleic Acids, 2023, 1.
4′-チオ核酸によるセントラル ドグマへの挑戦
田良島 典子、南川 典昭、核酸医薬・mRNA医薬の製造分析の基礎と基盤技術開発、CMC出版
Synthesis of oligonucleotides containing 5′-homo-4′-selenouridine derivative and its increased resistance against nuclease
As technologies using RNA or DNA have been developed, various chemical modifications of nucleosides have been attempted to increase the stability of oligonucleotides. Since it is known that 2′-OMe-modification greatly contributes to increasing the stability of oligonucleotides, we added 2′-OMe to our previously developed 4′-selenonucleoside and 5′-homo-4′-selenonucleoside as the modified monomers for oligonucleotide: 2′-methoxy-4′-selenouridine (2′-OMe-4′-Se-U) and 5′-homo-2′-methoxy-4′-selenouridine (5′-homo-2′-OMe-4′-Se-U). We synthesized oligonucleotides containing the chemically modified 4′-selenouridine and evaluated their thermal stability and nuclease resistance. In conclusion, the nuclease stability of the oligonucleotide containing 5′-homo-2′-OMe-4′-selenouridine increased while its thermal stability decreased.
Jinha Yu, Ji Won Kim, Girish Chandra, Noriko Saito–Tarashima et al., Bioorg. Med. Chem. Lett. 2022, 87, 129172.

Synthesis and properties of fully-modified 4′-selenoRNA, an endonuclease-resistant RNA analog
A large number of chemically modified oligonucleotides (ONs) have been developed for RNA-based technologies. In most modified RNAs, the characteristic 2′-hydroxyl (2′-OH) groups are removed to enhance both nuclease resistance and hybridization ability. However, the importance of the 2′-OH group in RNA structure and function is well known. Here, we report the synthesis and properties of 4′-selenoRNA in which all four nucleoside units retain the 2′-OH groups but contain a selenium atom instead of an oxygen atom at the 4′-position of the furanose ring. 4′-SelenoRNA has enhanced ability to form duplexes with RNA, and high endonuclease resistance despite the presence of the 2′-OH groups. X-ray crystallography analysis showed that the 4′-selenoRNA duplex adopts an A-conformation, similar to natural RNA, although one 4′-selenocytidine residue has unusual South-type sugar puckering. Furthermore, preliminary studies using 4′-seleno-modified siRNAs suggest that 4′-selenoRNA may be applicable to RNA interference technology. Collectively, our results raise the possibility of a new class of modified RNA in which 2′-OH groups do not need to be masked.
Masashi Ota et al., Bioorg. Med. Chem. 2022, 76, 117093.

2022
The Golgi-resident protein ACBD3 concentrates STING at ER-Golgi contact sites to drive export from the ER
STING, an endoplasmic reticulum (ER)-resident receptor for cyclic di-nucleotides (CDNs), is essential for innate immune responses. Upon CDN binding, STING moves from the ER to the Golgi, where it activates downstream type-I interferon (IFN) signaling. General cargo proteins exit from the ER via concentration at ER exit sites. However, the mechanism of STING concentration is poorly understood. Here, we visualize the ER exit sites of STING by blocking its transport at low temperature or by live-cell imaging with the cell-permeable ligand bis-pivSATE-2′F-c-di-dAMP, which we have developed. After ligand binding, STING forms punctate foci at non-canonical ER exit sites. Unbiased proteomic screens and super-resolution microscopy show that the Golgi-resident protein ACBD3/GCP60 recognizes and concentrates ligand-bound STING at specialized ER-Golgi contact sites. Depletion of ACBD3 impairs STING ER-to-Golgi trafficking and type-I IFN responses. Our results identify the ACBD3-mediated non-canonical cargo concentration system that drives the ER exit of STING.
Kou Motani, Noriko Saito–Tarashima et al., Cell Rep. 2022, 41, 111868.

Cas9-mediated DNA cleavage guided by enzymatically prepared
4′-thio-modified RNA
CRISPR-Cas9-mediated DNA editing relies on guide RNAs (gRNAs) that direct site-specific DNA cleavage by the Cas endonuclease. Because natural gRNA is susceptible to intracellular degradation, it is desirable to chemically protect it for efficient editing. Using 4′-thioribonucleoside 5′-triphosphates and T7 transcription, we have prepared 4′-thio-modified gRNAs that guide Cas9-mediated DNA cleavage. This approach is a simple way to obtain chemically modified RNA suitable for CRISPR-Cas9 DNA editing.
Noriko Saito–Tarashima et al., Org. Biomol. Chem. 2022, 20, 5245–5248.

4′-チオRNAにより構成される環状ジヌクレオチドアナログの創製
Noriko Saito–Tarashima et al., 日本ケミカルバイオロジー学会誌 2022, vol. 15
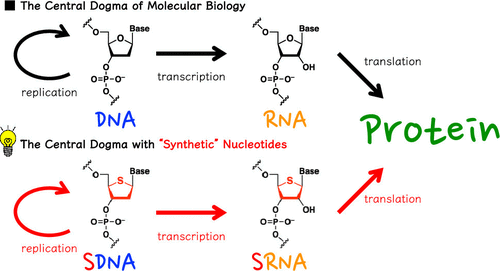
Rewriting the Central Dogma with Synthetic Genetic Polymers
DNA and RNA are ubiquitous molecules responsible for storage and transmission of genetic information and together comprise the central dogma of molecular biology. However, the recent emergence of synthetic genetic polymers is providing an opportunity to challenge the fundamental principles of life. Herein, we describe the ongoing attempts to rewrite the central dogma with 4′-thioDNA and 4′-thioRNA, which feature a sulfur instead of an oxygen atom in the furanose ring moiety. Using reconstituted Escherichia coli gene expression machinery, studies have shown that the genetic information conserved in 4′-thioDNA can be transcribed to 4′-thioRNA and eventually translated into protein, mirroring the processes that occur in nature. Such studies underscore the feasibility of controlling life by substances other than DNA and RNA.
Noriko Saito–Tarashima et al., Chem. Pharm. Bull. 2022, 70, 310–315.

Synthesis and Anti-dengue Virus Activity of 5-Ethynylimidazole-4-carboxamide (EICA) Nucleotide Prodrugs
We previously showed that 5-ethynyl-(1-β-D-ribofuranosyl)imidazole-4-carboxamide (1; EICAR) is a potent anti-dengue virus (DENV) compound but is cytotoxic to some cell lines, while its 4-thio derivative, 5-ethynyl-(4-thio-1-β-D-ribofuranosyl)imidazole-4-carboxamide (2; 4′-thioEICAR), has less cytotoxicity but also less anti-DENV activity. Based on the hypothesis that the lower anti-DENV activity of 2 is due to reduced susceptibility to phosphorylation by cellular kinase(s), we investigated whether a monophosphate prodrug of 2 can improve its activity. Here, we first prepared two types of prodrug of 1, which revealed that the S-acyl-2-thioethyl (SATE) prodrug had stronger anti-DENV activity than the aryloxyphosphoramidate (so-called ProTide) prodrug. Based on these findings, we next prepared the SATE prodrug of 4′-thioEICAR 18. As expected, the resulting 18 showed potent anti-DENV activity, which was comparable to that of 1; however, its cytotoxicity was also increased relative to 2. Our findings suggest that prodrugs of 4′-thioribonucleoside derivatives such as EICAR (1) represent an effective approach to developing potent biologically active compounds; however, the balance between antiviral activity and cytotoxicity remains to be addressed.
Motoki Nakamura et al., Chem. Pharm. Bull. 2022, 70, 220-225.

2021
Convenient Synthesis of 3-Deazapurine Nucleosides (3-Deazainosine, 3-Deazaadenosine and 3-Deazaguanosine) Using Inosine as a Starting Material
A convenient synthetic method for preparing 3-deazapurine nucleosides (3-deazainosine, 3-deazaadenosine, and 3-deazaguanosine) from inosine via a 5-ethynyl-1-β-D-ribofuranosylimidazole-4-carboxamide (EICAR) derivative, which is a key intermediate, is described. A large-scale synthesis of an EICAR derivative starting from inosine was achieved in six steps via dinitrophenylation at the N1 position followed by ring opening, iodination of the resulting 5-amino group, and a palladium-catalyzed cross-coupling reaction. The resulting EICAR derivative was then converted into 3-deazainosine, 3-deazaadenosine, and 3-deazaguanosine. This route enabled us to synthesize 3-deazapurine nucleosides conveniently in good yields.
Naoto Hinotani et al., Curr. Protoc. Nucleic Acid Chem. 2021, 11, e297.
Synthesis of a Cyclic Dinucleotide Analogue with Ambiguous Bases, 5-Aminoimidazole-4-carboxamide
Cyclic dinucleotides (CDNs) are second messengers composed of two purine nucleotides. In recent years, the structural diversity of CDNs and their functionality in biological processes are being intensely studied. Herein we report the chemical synthesis of cyclic di-5-aminoimidazole-4-carboxamide-1-β-d-ribofuranosyl monophosphate (c-di-ZMP) (1), which consists of two 5-amino-4-imidazolecarboxamide ribonucleotides (Z-ribonucleotides) linked via two phosphodiester linkages. Construction of the CDN skeleton with an N1-dinitrophenylhypoxanthine base (HxaDNP-base) by phosphoramidite chemistry and the subsequent ring-opening reaction of HxaDNP-base successfully yielded the desired 1.
Noriko Saito–Tarashima et al., J. Org. Chem. 2021, 86, 15004–15040.

5-Hydroxymethyltubercidin Exhibits Potent Antiviral Activity against Flaviviruses and Coronaviruses, including SARS-CoV-2
Newly emerging or re-emerging viral infections continue to cause significant morbidity and mortality every year worldwide, resulting in serious effects on both health and the global economy. Despite significant drug discovery research against dengue viruses (DENVs) and severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), no fully effective and specific drugs directed against these viruses have been discovered. Here, we examined the anti-DENV activity of tubercidin derivatives from a compound library from Hokkaido University and demonstrated that 5-hydroxymethyltubercidin (HMTU, HUP1108) possessed both potent anti-flavivirus and anti-coronavirus activities at submicromolar levels without significant cytotoxicity. Furthermore, HMTU inhibited viral RNA replication and specifically inhibited replication at the late stages of the SARS-CoV-2 infection process. Finally, we demonstrated that HMTU 5′-triphosphate inhibited RNA extension catalyzed by the viral RNA-dependent RNA polymerase. Our findings suggest that HMTU has the potential of serving as a lead compound for the development of a broad spectrum of antiviral agents, including SARS-CoV-2.
Kentaro Uemura et al., iScience 2021, 24, 103120.

Replacement of oxygen with sulfur on the furanose ring of
cyclic dinucleotides enhances the immunostimulatory effect
via STING activation.
Cyclic dinucleotides (CDNs) are secondary messengers composed of two purine nucleotides linked via two phosphodiester linkages: c-di-GMP, c-di-AMP, 3′,3′-cGAMP, and 2′,3′-cGAMP. CDNs activate the stimulator of interferon genes (STING) and trigger immune responses in mammalian species. CDNs are thus fascinating molecules as drug candidates, and chemically stable CDN analogues that act as STING agonists are highly desired at present. We herein report the practical synthesis of 4′-thiomodified c-di-AMP analogues, which have sulfur atoms at the 4′-position on the furanose ring instead of oxygen atoms, using simple phosphoramidite chemistry. The resulting 4′-thiomodified c-di-AMP analogues acted as potent STING agonists with long-term activity. Our results show that replacing O4′ on CDNs with sulfur can lead to enhanced immunostimulatory effects via STING activation.
Noriko Saito–Tarashima et al., RSC Med. Chem. 2021, 12, 1519–1524.

Synthesis and Properties of 4′-ThioLNA/BNA
To develop a new nucleoside analogue applicable to oligonucleotide therapeutics, we designed a 4′-thio analogue of an LNA/BNA monomer. Synthesis of 4′-hydroxymethyl-4′-thioribonucleoside was achieved by a tandem ring-contraction-aldol reaction of a 5-thiopyranose derivative and the subsequent Pummerer-type thioglycosylation reaction of the corresponding sulfoxide. Treatment of 4′-hydroxymethyl-4′-thiopyrimidine nucleosides with diphenyl carbonate in the presence of catalytic NaHCO3 gave the desired 4′-thioLNA/BNA monomers, which were introduced into oligonucleotides.
Rion Maeda et al., Org. Lett. 2021, 10, 4062–4066.

2020
Gene expression of 4′-thioguanine DNA via 4′-thiocytosine RNA
DNA and RNA nucleotides are ubiquitous molecules that store and transmit genetic information. The emergence of synthetic elements that fulfill the function of DNA and RNA provides an alternative gene expression system. Herein, we demonstrate the gene expression of 4′-thioguanine DNA (dSG DNA) via 4′-thiocytosine RNA (dSC RNA) to give green fluorescent protein (GFPuv) in a single test tube. In replication, transcription, and translation, DNA/RNA polymerases and Escherichia coli (E. coli) ribosome can tolerate the replacement of O4′ with S4′ in the nucleotide, despite the fact that sulfur has a larger atomic radius than oxygen. Additionally, dSG DNA and dSC RNA acted as alternative genetic polymers to natural DNA and RNA for protein synthesis in artificial cells comprising a reconstituted E. coli gene expression machinery. This work involved simple experiments that are widely used in molecular biology, but which underscore the feasibility of life control by substances other than DNA/RNA nucleotides.
Noriko Saito–Tarashima et al., J. Am. Chem. Soc. 2020, 142, 17255–17259.
A unique gene-silencing approach, using an intelligent RNA expression device (iRed), results in minimal immune stimulation when given by local intrapleural injection in malignant pleural mesothelioma
Background: We have recently introduced an intelligent RNA expression device (iRed), comprising the minimum essential components needed to transcribe short hairpin RNA (shRNA) in cells. Use of iRed efficiently produced shRNA molecules after transfection into cells and alleviated the innate immune stimulation following intravenous injection. Methods: To study the usefulness of iRed for local injection, the engineered iRed encoding luciferase shRNA (Luc iRed), complexed with cationic liposomes (Luc iRed/liposome-complexes), was intrapleurally injected into an orthotopic mesothelioma mouse model. Results: Luc iRed/liposome-complexes markedly suppressed the expression of a luciferase marker gene in pleurally disseminated mesothelioma cells. The suppressive efficiency was correlated with the expression level of shRNA within the mesothelioma cells. In addition, intrapleural injection of iRed/liposome-complexes did not induce IL-6 production in the pleural space and consequently in the blood compartment, although plasmid DNA (pDNA) or dsDNA (the natural construct for iRed) in the formulation did. Conclusion: Local delivery of iRed could augment the in vivo gene silencing effect without eliciting pronounced innate immune stimulation. Our results might hold promise for widespread utilization of iRed as an RNAi-based therapeutic for intracelial malignant cancers.
Hidenori Ando et al., Molecules. 2020, 25, 1725.
フラノース環酸素原子をイオウ、セレン原子に置換した核酸誘導体の有機合成化学
To date, a number of chemically modified oligonucleotides (ONs) have been designed for use in nucleic acids-based therapeutics. In our group, we have been intensely working on the synthesis of 4’-thio and 4’-seleno ONs having sulfur or selenium atoms in place of furanose ring oxygen. To prepare 4’-thio and 4’-seleno ribonucleosides, we applied the Pummerer reaction of 4-thio or 4-seleno sugar with nucleobases, and the resulting 4’-thio and 4’-seleno ribonucleoside units were incorporated into ONs. Under the standard phosphoramidite conditions, 4’-thioRNA was obtained in good yields. While 4’-selnoRNA was given in very low yields. We carefully investigated ON synthesis containing 4’-selenoribonucleosides under standard phosphoramidite conditions. As a result, we found the unexpected strand-break occurred during oxidation step using I2. On the basis of this finding, we succeeded in the first synthesis of a fully modified 4’-selenoRNA by using tert-butyl hydroperoxide as an alternative oxidant.
Masashi Ota et al., 有機合成化学協会誌 2020, 78, 446–455.